
研究揭示肠癌驱动脂质合成促肿瘤进展机制
中山大学肿瘤防治中心鞠怀强教授、徐瑞华院士与南方医科大学李博教授等研究发现,代谢酶AHCY与其代谢产物腺苷(ADO) 形成的AHCY-ADO复合物,能够通过一种非经典途径,特异性调控mRNA的N6-甲基腺苷(m⁶A)水平。该复合物通过驱动AHCY二聚化,增强其与RNA去甲基化酶FTO的结合,从而特异性地阻碍FTO对脂肪合成关键基因的mRNA上m⁶A修饰的清除。这导致脂质合成通路被持续激活,为肿瘤细胞增殖提供充足的代谢原料。研究者基于该机制设计了一种肽,通过破坏AHCY二聚化实现有效抑制小鼠肿瘤生长。(Cell Res. 2026, 36: 152-172.)
(2)-86.jpg")
肿瘤的发生与发展始终伴随着复杂的代谢重编程,代谢与表观遗传之间的交互对话是研究热点。甲硫氨酸循环是细胞内连接代谢与表观遗传的核心枢纽,其终产物S-腺苷甲硫氨酸(SAM)作为一种通用的甲基供体,参与包括m⁶A在内的RNA、DNA和组蛋白的甲基化修饰反应,影响基因表达调控网络。
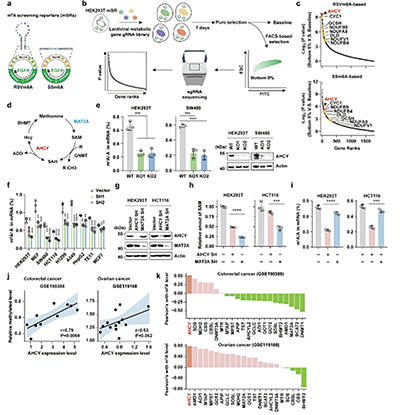
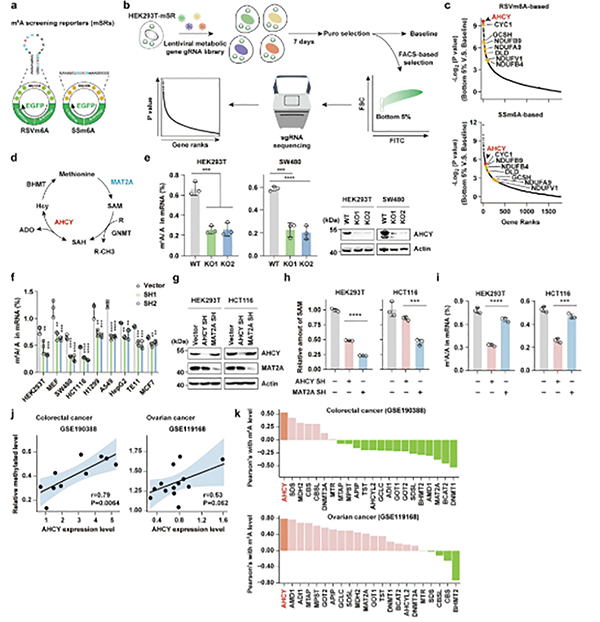
研究者设计了一套筛选系统,将一段可被m⁶A修饰的特异性序列(GGACU)嵌入环状RNA中,该序列的甲基化状态直接决定下游绿色荧光蛋白(GFP)的表达水平——m⁶A水平越高,荧光信号越强。将该系统与覆盖1700余个代谢基因的CRISPR-Cas9敲除文库相结合,便可通过筛选荧光信号异常的细胞,高通量鉴定调控m⁶A的代谢因子。
筛选结果锁定了AHCY,甲硫氨酸循环中的一个关键酶,负责催化S-腺苷同型半胱氨酸(SAH)可逆性水解为腺苷(ADO)和同型半胱氨酸。MAT2A等其他甲硫氨酸循环关键酶并未在筛选中显著富集,提示AHCY可能以独立于甲硫氨酸循环或SAM的方式调控m⁶A修饰。
敲低或敲除AHCY能显著降低结直肠癌(SW480、HCT116)等多种肿瘤细胞中的m⁶A水平,而过表达AHCY则能使m⁶A水平升高。在AHCY缺失的细胞中,补充甲硫氨酸循环的多种代谢物(包括SAM)并不能完全挽救m⁶A水平的下降,而单独补充AHCY的代谢产物ADO却能显著提升多种细胞系的m⁶A水平。MAT2A作为直接催化SAM合成的酶,其缺失导致细胞内SAM水平严重下滑,m⁶A的水平下调却在AHCY缺失细胞中更显著,提示AHCY存在SAM非依赖的调控功能。
分子机制探索研究显示,AHCY能通过与其代谢产物ADO结合,形成稳定的AHCY-ADO复合物。该复合物诱导AHCY形成二聚体,二聚化AHCY与RNA去甲基化酶FTO直接结合,从而抑制FTO活性。FTO活性被抑制后,其负责清除mRNA上m⁶A修饰的功能便会受阻。
(2)-87.jpg")
富集分析显示,基因m⁶A修饰被特异性保护的是脂肪酸生物合成通路。关键限速酶乙酰辅酶A羧化酶1(ACACA) 和去饱和酶硬脂酰辅酶A去饱和酶1(SCD1) 的mRNA受AHCY-ADO/FTO通路的调控最为显著。AHCY-ADO复合物通过降低FTO的去甲基化活性,提高ACACA和SCD1转录本上的m⁶A修饰水平,增强其稳定性和表达,从而促进肿瘤细胞的脂肪酸从头合成与脂滴积累。
结直肠癌小鼠实验结果显示,肠道特异性敲除AHCY能够显著抑制肿瘤生长。临床样本分析显示,在结直肠癌和肺癌等肿瘤组织中,AHCY蛋白表达与脂肪酸合成酶ACC1的表达呈正相关,而与FTO表达呈负相关,且高表达AHCY的患者总体生存期更短。
研究者基于AHCY二聚体界面设计了一种干扰肽(AA #7),该肽能有效破坏AHCY二聚化,抑制其与FTO的相互作用,并在体外细胞实验和小鼠体内实验中有效抑制肿瘤细胞增殖和生长,显示靶向这一新机制的治疗潜力。
该研究深化了我们对肿瘤代谢网络的理解,揭示AHCY-ADO-FTO-m⁶A通路可作为一个有前景的肿瘤代谢治疗靶点。针对AHCY这一机制的干预策略,如优化AA #7类似物或寻找小分子抑制剂,有望成为切断肿瘤供应从而遏制其生长的疗法。
(编译 张媛媛)











